Na jaře příštího roku předloží Evropská komise novelizaci čtyř klíčových předpisů upravujících pravidla lékového trhu EU. Co má či nemá obsahovat, to je předmětem značných diskuzí. Shoda panuje na tom, že je potřeba minimálně urychlit schvalovací procesy a posílit expertní zázemí lékových agentur. Nejde ale jen o lékovou legislativu. Velká očekávání vyvolává i příprava společného hodnocení zdravotnických technologií (HTA) a vznik evropského prostoru pro sdílení zdravotnických dat, jak zaznělo minulý minulý týden v Bruselu na konferenci Farmaceutická strategie pro Evropu: implementace, úspěchy, výzvy.
Evropský lékový trh čeká rozsáhlá změna pravidel, s konkrétním návrhem by měla Evropská komise přijít na jaře příštího roku. Jak nedávno popsala eurokomisařka pro zdraví Stella Kyriakidesová v rozhovoru pro Zdravotnický deník, cílem této novelizace je podpořit evropský výzkum a vývoj inovativních, kvalitních, účinných a bezpečných léků, zejména v oblastech, kde jsou nejvíce potřeba, a ty učinit snadněji dostupné a přístupné v celé EU.
Není divu, že očekávání jsou veliká a vyvolávají značné diskuze o tom, co by návrhy měly obsahovat a co nikoli, jak minulý týden ukázala konference Farmaceutická strategie pro Evropu: implementace, úspěchy, výzvy, která se konala na půdě Stálého zastoupení ČR při EU v Bruselu.

„To, co nás čeká, je opravdu velká změna. Abychom se dopracovali k opravdu dobrému návrhu, musíme velmi dobře poznat a pochopit, jak současný systém funguje, co je v něm důležité a jak je to upraveno legislativně,“ připomněla ve svém projevu na konferenci ředitelka Evropské federace farmaceutického průmyslu a asociací (EFPIA) pro strategii v oblasti regulace Sini Eskola.
Zrychlit hodnocení, najít experty
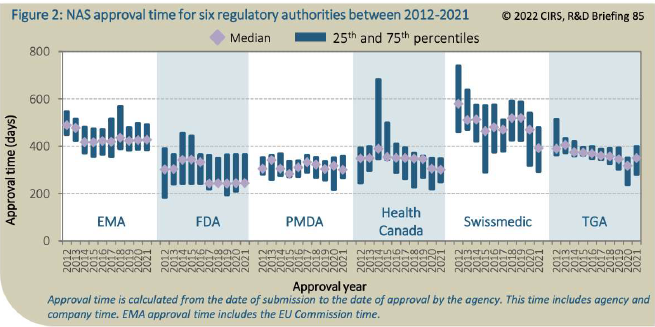
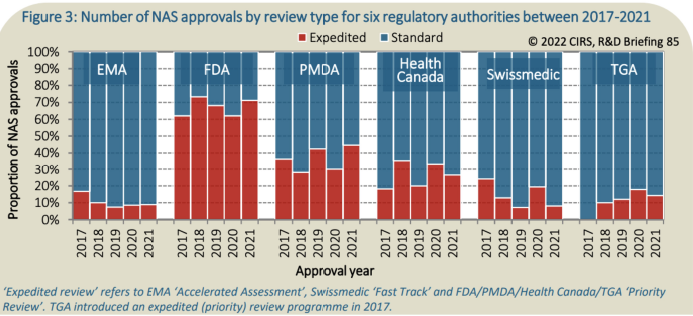
EFPIA letos v lednu zveřejnila po dvou letech přezkumu a interních diskuzí vlastní analýzu současného legislativního nastavení lékové trhu v Evropské unii a jeho silných a slabých stránek. Podle tohoto dokumentu je zapotřebí zejména zrychlit hodnotící a schvalovací procesy, počínaje hodnocením žádosti o registraci inovativního léčivého přípravku, což se v Evropské unii děje centralizovaně a je to úkolem Evropské lékové agentury (EMA). Jak na konferenci upozornila šéfka EFPIA Nathalie Mollová, posuzování těchto žádostí trvalo v roce 2021 agentuře EMA v průměru dvakrát déle než v USA (428 dní vs. 245 dní). Navíc v porovnání se státy jako USA, Kanada či Švýcarsko EMA také v mnohem menší míře využívá možností zrychleného hodnocení například u zvláště inovativních a potřebných přípravků.
Evropa dále potřebuje posílit svoji expertízu, pokračuje Eskola, to jak na straně agentury EMA, tak národních lékových agentur. „Potřebujeme se zamyslet nad tím, kde budeme shánět experty na nové technologie, bude zapotřebí například vyřešit otázku střetu zájmů,“ uvedla s tím, že průmysl je připraven platit vyšší poplatky, pokud tyto peníze půjdou na zajištění potřebných kapacit.
Nejde totiž již jen o hodnocení léčivého přípravku v předregistrační fázi. Inovativní moderní terapie – typické je to v oblasti vzácných onemocnění s malými skupinami pacientů nebo u individuálně připravovaných genových terapií – vstupují do registračního procesu s omezeným množstvím dat. A tyto případy budou s ohledem na současné technologické trendy stále častější. Proces hodnocení přípravku tak bude muset pokračovat i po schválení registrace, jak začnou postupně přicházet data z reálné klinické praxe.
Pozornost je dále podle Sini Eskola věnovat i kombinovaným přípravkům, které se skládají z léku a jednoho nebo více zdravotnických prostředků. „V současné době tvoří kolem čtvrtiny inovativních přípravků, nejde tedy o žádnou okrajovou záležitost,“ upozorňuje ředitelka. Léky a zdravotnické prostředky však procházejí zcela odlišným hodnotícím a schvalovacím procesem, a to vytváří značnou míru nejistoty. „Měla by vzniknout definice kombinovaného léčivého přípravku, abychom dosáhli větší předvídatelnosti systému. Měli bychom definovat odpovědnosti jednotlivých aktérů v systému,“ apeluje.
Velké zjednodušení by podle ní také přinesla elektronizace příbalových letáků.
Mohlo by vás zajímat


Společné HTA je příležitostí, kterou lze stále promarnit
Nejde však je o novou lékovou legislativu, která by měla pomoci uskutečnit vizi komisařky Kyriakidesové popsanou v úvodu článku. Naděje se vkládají také do společného unijního hodnocení zdravotnických technologií (HTA), procesu, který by měl spojit odborné kapacity států EU a sdílet velmi náročné hodnocení účinnosti a bezpečnosti moderních terapií. Od roku 2025 by takto měly být hodnoceny inovativní přípravky určené pro onkologické diagnózy, o tři roky později léky pro vzácná onemocnění a od roku 2030 pak všechny všechny léčivé přípravky registrované centralizovanou procedurou, tedy agenturou EMA.
Jak ale upozorňuje Ansgar Hebborn, vedoucí pracovní skupiny EFPIA pro hodnocení zdravotnických technologií ze společnosti Roche, v současné době se teprve připravují společná prováděcí pravidla a metodika. A ta teprve prokáží, zda unijní HTA bude skutečně efektivní. Nemalou roli bude hrát skutečnost, že členské státy jsou na různé úrovni hodnocení HTA a tam, kde s principy HTA již nějakým způsobem pracují, to vypadá vždy trochu jinak než jinde. Požadavky na data a dokumentaci od výrobců se liší stát od státu. „Na stejná klinická data jsou aplikována různá kritéria,“ upozorňuje Hebborn. Je proto velmi těžké vygenerovat potřebná data tak, aby vyhovovala všem.

„Nařízení (EU) o HTA je velkou příležitostí, která může leccos vyřešit. Zároveň ale ponechává značný prostor pro členské státy a další aktéry, aby z toho vytvořili buď úspěšný, nebo také neúspěšný systém,“ varuje Hebborn. Podle něj nařízení zakotvuje jen opravdu základní pravidla a mnohé podrobnosti ponechává na prováděcí předpisy a metodiku. Bude tedy zapotřebí dohlédnout na to, aby do roku 2025 byl systém nastaven tak, že povede k očekávaným výsledkům. „A to se neobejde bez skutečně aktivních konzultací i s průmyslem a výrobci,“ připomíná.
Navíc teprve až praxe ukáže, jak s výstupy společného HTA naloží samotné členské státy. Především kvůli nim celý systém vznikl, i když výsledky hodnotících zpráv nakonec pro ně nebudou závazné. „Je na čase, aby se státy samotné zamyslely, jak k tomu přistoupí. Zda upraví své národní procesy tak, aby i jim společné HTA přineslo užitek. A tohle všechno musí být hotové do tří let,“ uzavírá Ansgar Hebborn.
Sdílení zdravotnických dat: mnoho nadějí i obav
Hodnocení moderních inovativních terapií se, jak již bylo zmíněno, často pojí s menšími skupinami pacientů, a tedy menším objemem dostupných informací ve chvíli registrace a větší mírou nejistoty pro národní lékové agentury a plátce. Zapojení dat z konkrétní klinické praxe a jejich následné vyhodnocování se tak do budoucna stane v takových případech nezbytností. „A řešení může přinést společný evropský prostor pro sdílení zdravotnických dat,“ je přesvědčený náměstek ministra zdravotnictví Jakub Dvořáček.
Návrh nařízení, který právě takový prostor zakládá, předložila Evropská komise letos v květnu. Podle Dvořáčka tento virtuální prostor, díky němž budou moci ministerstva, lékové orgány, vědecké instituce, univerzity nebo i výrobci za přísně definovaných podmínek sdílet zdravotnické údaje z celé EU, nabízí odpověď na řadu současných otázek.

„Bude mnohem jednodušší získat data z klinické praxe, porovnat dostupnost a úhrady v jednotlivých zemích, obhájit náklady na léčbu, získat podklady pro přípravu legislativy, vyhodnotit skutečnou efektivitu společných nákupů,“ vypočítává Dvořáček některé z očekávaných benefitů. Pokud chce Evropa zůstat globálně konkurenceschopná, tvrdí, musí mít stejné možnosti, jak se rychle dostat k datům, jako tomu je v USA či jiných zemích.
Na druhou stranu je nutné si přiznat, že návrh vyvolává mnoho emocí na všech stranách, připouští dále náměstek. „Lékaři mají obavy z interpretace dat, pacienti řeší ochranu soukromí a proces anonymizace, průmysl dopady na cenové a úhradové mechanismy a státy, kde seženou peníze na všechny ty změny,“ jmenuje Dvořáček hlavní obavy, které doposud při projednávání návrhu zazněly.
Bez diskuze určitě nezůstane také nastavení kritérií, podle kterých lze o tato data požádat. „Současný návrh například vylučuje použití pro marketingové účely, ale víme, že průmysl chápe marketing šířeji,“ konstatuje náměstek. Bude tak podle něj nutné odlišit data pro „čistý“ marketing a data, která je skutečně nezbytné získat před vstupem na určitý trh.
Úkoly však čekají i na členské státy samotné co se týče kvality sdílených dat, upozorňuje ještě Ansgar Hebborn. „Pokud chceme znát a sdílet skutečné výsledky určité terapie, musíme je ale také dokázat změřit. Nejde totiž jen o přípravek samotný. Jde i o lékaře, pacienty, jak jsou proškolení, jak se lék podává a podobně,“ zdůraznil.
Návrh nařízení nyní projednávají poslanci v Evropském parlamentu a členské státy v Radě EU. Obě unijní instituce se musí nakonec shodnout na společném kompromisním textu, a to může trvat ještě řadu měsíců.
Helena Sedláčková
Foto: Dagmar Kneřová
Konferenci Farmaceutická strategie pro Evropu: implementace, úspěchy, výzvy organizovala Asociace inovativního farmaceutického průmyslu (AIFP) pod záštitou českého předsednictví v Radě EU. Záznam celé akce můžete sledovat zde.