Příští rok otřese trhem se zdravotnickými prostředky. Výrobci totiž budou muset (mimo jiné) požádat o novou certifikaci dle evropského nařízení o zdravotnických prostředcích MDR a následně na certifikaci uzavřít smlouvu s oznámeným subjektem (čili, postaru, s notifikovanou osobou) podle MDR. Zatímco doposud tak šlo o sdílený problém, brzy se ukáže, zda se výrobci na nová, přísná pravidla připravovali a podaří se jim obstát. Aktuální stav přiblížil Zdravotnickému deníku právní expert ze vzdělávací a poradenské společnosti Porta Medica Jakub Král.
Kevropským nařízením MDR s IVDR (nařízení regulující uvádění zdravotnických prostředků pro diagnostiku in vitro) se i po jejich schválení přijímají další opatření a dokumenty. Došlo letos k nějakým změnám u těchto nařízení?
Ano. V prvním čtvrtletí letošního roku bylo schváleno novelizační nařízení (EU) 2023/607, které některým výrobcům prodloužilo přechodné období, po které mohou na trh uvádět tzv. legacy devices, tedy zdravotnické prostředky, u nichž byla shoda s požadavky EU posouzena ještě dle dřívějších směrnic MDD nebo AIMDD. Prodloužení přechodných období je však striktně podmíněno včasným naplněním mnoha požadavků. Aktuálně se diskutuje především o tom, jak splnění všech podmínek prokázat vůči dozorovým orgánům i obchodním partnerům. Není snadné vysvětlovat a dokládat, že platnost certifikátu sice již vypršela, avšak s ohledem na splnění specifických požadavků nařízení 2023/607 došlo ex lege k prodloužení nebo obnovení platnosti certifikátu.
Zároveň byla v MDR i v IVDR odstraněna roční lhůta pro doprodej legacy devices řádně uvedených na trh. Nově je tedy možné tyto produkty prodávat a uvádět do provozu až do doby expirace, pokud je výrobcem stanovena, v ostatních případech bez časového omezení.
Můžete připomenout, co už v tuto chvíli výrobci podle nařízení musejí a jaké jsou nejbližší termíny, kdy přibudou další povinnosti?
Mohlo by vás zajímat

Podle MDR a IVDR musí již dnes kompletně postupovat výrobci nových prostředků, neboť ti nemohou využít status legacy devices. Zároveň musí MDR plně implementovat všichni výrobci nesterilních zdravotnických prostředků rizikové třídy I bez měřicí funkce, pokud tyto prostředky mají stejnou klasifikaci dle MDD i MDR. IVDR pak musí zcela splňovat výrobci nesterilních IVD prostředků, které nově spadají do třídy A.
Nejbližší termín, který otřese s trhem zdravotnických prostředků, představuje příští rok, konkrétně květen a září 2024, kdy budou muset výrobci legacy devices splnit hned několik podmínek. Například budou muset mít již zavedený systém řízení kvality dle MDR, budou muset požádat o novou certifikaci dle MDR a následně uzavřít smlouvu na tuto certifikaci s oznámeným subjektem (notifikovanou osobou) podle MDR.
Jak jsou na tom v tuto chvíli výrobci, zvládají nové požadavky plnit?
Pro výrobce teď přichází nejobtížnější období – chvíle pravdy, protože teď už je to jen na nich a na jejich připravenosti. Doposud to byl sdílený problém. Neustále se poukazovalo na nedostatečné certifikační kapacity, absenci prováděcích předpisů, chybějící výkladové MDCG dokumenty, nevydané společné specifikace, absentující harmonizované technické normy a podobně. To všechno dnes již odpadá. Výrobci nyní musí předvést výsledky své šestileté práce na plnění požadavků MDR.
Jak to vypadá se certifikačními kapacitami? Už je dostatek oznámených subjektů dle MDR a IVDR?
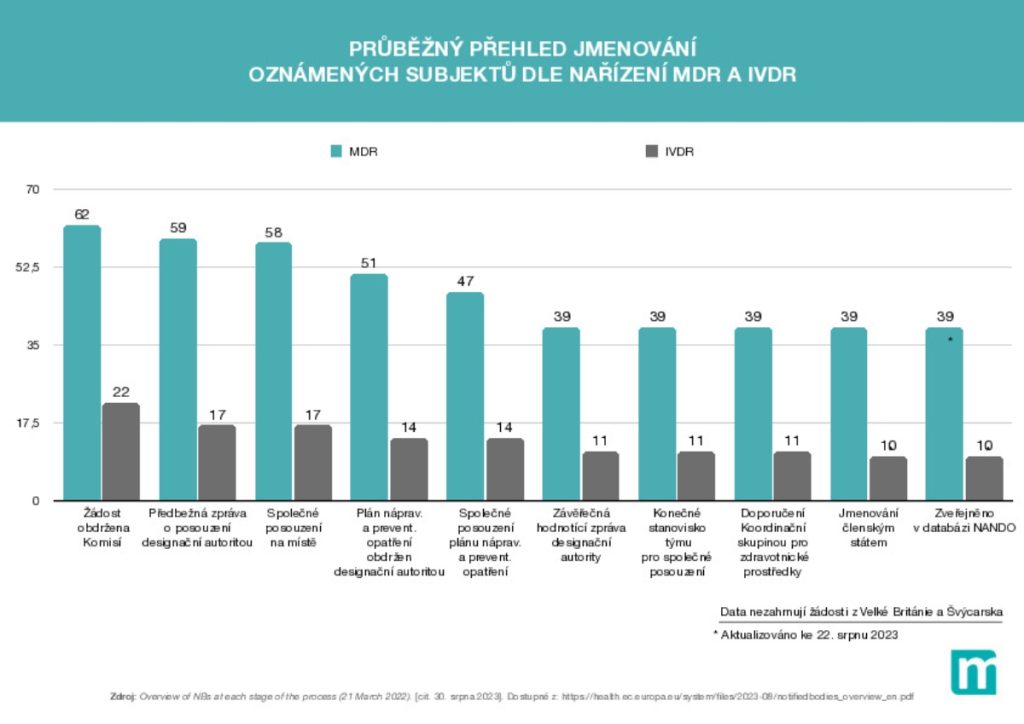
Situace se pomalu zlepšuje a stabilizuje. Aktuálně působí 39 oznámených subjektů dle MDR a 10 oznámených subjektů dle IVDR. Nicméně v procesu posuzování je dalších 23 žadatelů o tento status dle MDR včetně Českého metrologického institutu a 12 žadatelů dle IVDR. U obecných prostředků už tedy přestává platit argument, že by nebylo kde certifikovat. Větší počet oznámených subjektů navíc povede ve střednědobém horizontu k tolik potřebné konkurenci, což bude mít za následek i pro výrobce příznivější cenovou politiku. Dosud si totiž nemohli moc vybírat a byli rádi, když je vůbec někdo „vzal“. V oblasti IVD to tak pozitivně nehodnotím, protože tam bude nezbytné nově certifikovat obrovské množství prostředků, které doposud certifikaci nepodléhaly. V tomto segmentu jsou dle mého názoru certifikační kapacity i nadále nedostatečné.
Kde teď vnímáte největší problém, jak se s požadavky vypořádávají např. výrobci individuálně zhotovovaných prostředků (prostředků na zakázku), které jste zmiňoval jako jedny z nejohroženějších v minulosti?
Tady se bohužel mé obavy potvrzují. Mnoho z těchto výrobců doposud nedisponuje vyhovující technickou dokumentací, nemají zavedený adekvátní systém řízení kvality a mnohdy ani neustanovili osobu odpovědnou za dodržování právních předpisů s potřebnou odbornou kvalifikací. Co se naopak výrazně zlepšilo, je podpora těchto výrobců ze strany SÚKL a MDCG. Vznikl celoevropský výkladový dokument, který podrobně vysvětluje, jak mají tito výrobci postupovat. SÚKL pro ně uspořádal i několik edukativních seminářů. Také mají výhodu, že s výjimkou implantabilních prostředků na zakázku spadajících do rizikové třídy III nepodléhají náročnému procesu certifikace. Situace v terénu však stále není dobrá a z postupu některých výrobců je zřejmé, že dokud nepřejde SÚKL od edukace k represi (udílení pokut za porušování právních předpisů), tito výrobci se o poctivou implementaci požadavků MDR ani nepokusí. Staví se k tomu ve stylu „kde není žalobce, není soudce“. Možná si to někteří vyhodnotili tak, že splnění nových požadavků se jim „nevyplatí“. Rozhodně to ale není tak, že by nové požadavky naplnit objektivně nešlo. Už máme zkušenost s několika opravdu malými výrobci de facto rodinného typu, kteří se s implementací MDR popasovali velmi solidně.
-mk-